
A medida que los semiconductores basados en silicio alcanzan sus límites de rendimiento, el nitruro de galio (GaN) se está convirtiendo en el material de referencia para avanzar en la tecnología LED. El inconveniente con el GaN es su gran cantidad de defectos. Entender mejor cómo se forman estos defectos a nivel atómico podría mejorar el rendimiento de los dispositivos fabricados con este material.
Esta degradación del material se debe a dislocaciones, cuando los átomos se desplazan en la estructura de la red cristalina. Cuando múltiples dislocaciones se mueven simultáneamente desde la fuerza de corte, las uniones a lo largo de los planos reticulares se estiran y eventualmente se rompen. A medida que los átomos se reordenan para reformar sus enlaces, algunos planos permanecen intactos mientras que otros se deforman permanentemente, con solo medio plano en su lugar. Si la fuerza de corte es lo suficientemente grande, la dislocación finalmente se extenderá a lo largo del borde del material.
La estratificación del GaN en diferentes sustratos hace que el problema sea mucho peor debido a que las estructuras reticulares generalmente no se alinean. Esta es la razón por la cual ampliar nuestra comprensión de cómo se forman los defectos del GaN a nivel atómico podría mejorar el rendimiento de los dispositivos fabricados con esta material.
Un equipo de investigadores ha dado un paso significativo hacia este objetivo al examinar y determinar seis configuraciones centrales de la red GaN. Presentaron sus resultados en el Journal of Applied Physics, de AIP Publishing.
“El objetivo es identificar, procesar y caracterizar estas dislocaciones para comprender completamente el impacto de los defectos en el GaN, de modo que podamos encontrar formas específicas de optimizar este material”, dijo Joseph Kioseoglou, investigador de la Universidad Aristóteles de Tesalónica y autor del artículo científico.
También hay problemas que son intrínsecos a las propiedades del GaN que producen efectos no deseados de cambio de color en la emisión de LED basados en GaN. Según Kioseoglou, esto podría abordarse potencialmente explotando diferentes orientaciones de crecimiento.
Análisis computacional mediante dinámicas moleculares
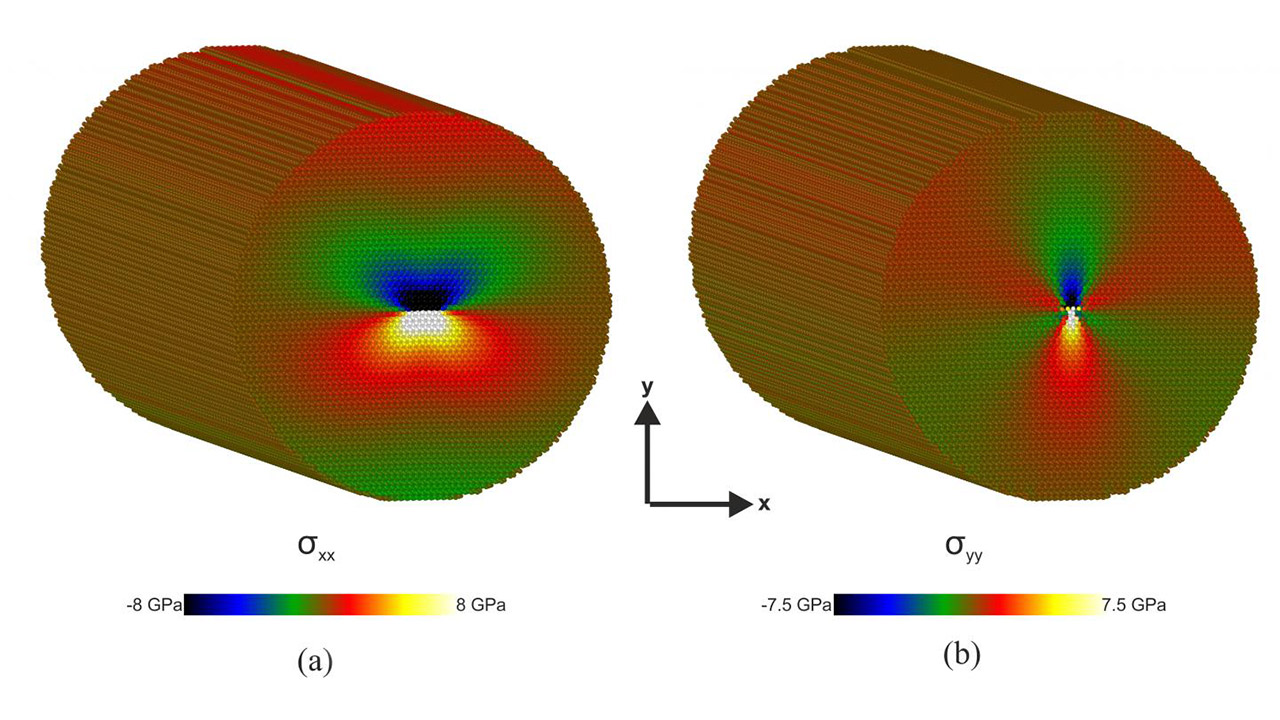
Los investigadores utilizaron el análisis computacional mediante dinámicas moleculares y simulaciones de la teoría funcional de la densidad para determinar las propiedades estructurales y electrónicas de las luxaciones del borde basal a lo largo de la dirección <1-100> en GaN. Las dislocaciones en esta dirección son comunes en las orientaciones de crecimiento semipolar.
El estudio se basó en tres modelos con diferentes configuraciones centrales. El primero consistía en tres átomos de nitrógeno (N) y un átomo de galio (Ga) para la polaridad Ga; el segundo tenía cuatro átomos de N y dos átomos de Ga; el tercero contenía dos átomos de N y dos átomos asociados con el núcleo de Ga. Los cálculos dinámicos moleculares se realizaron usando aproximadamente 15.000 átomos para cada configuración.
Los investigadores encontraron que las configuraciones de polaridad N exhibieron significativamente más estados en la “banda prohibida” en comparación con los de polaridad Ga, con las configuraciones N polar presentando valores de “banda prohibida” más pequeños.
«Existe una conexión entre los valores de banda prohibida más pequeños y la gran cantidad de estados dentro de ellos», dijo Kioseoglou. «Estos hallazgos potencialmente demuestran el papel del nitrógeno como un importante contribuyente a los efectos relacionados con la dislocación en dispositivos basados en GaN».
Créditos de portada: Physics Department, Aristotle University of Thessaloniki. “La imagen muestra las distribución de tensiones por átomo, (a) y (b) de las dislocaciones de eje a lo largo de la dirección <1-100> de la estructura cristalina Wurtzite del GaN”
